



Ocular tumours are rare but carry significant consequences. Choroidal melanoma occurs in approximately 6–10 per million people per year, with iris and conjunctival melanomas even less common at around 1 and 0.5 per million respectively1. These malignancies most commonly affect middle-aged and older individuals, particularly those with fair skin and light-coloured irides. While anterior tumours are often detected earlier, posterior lesions frequently remain asymptomatic and are therefore at greater risk of delayed diagnosis.
Although these conditions are uncommon and best managed in subspecialist centres, for both optometrists and ophthalmologists, early recognition and referral remain key to improving outcomes.
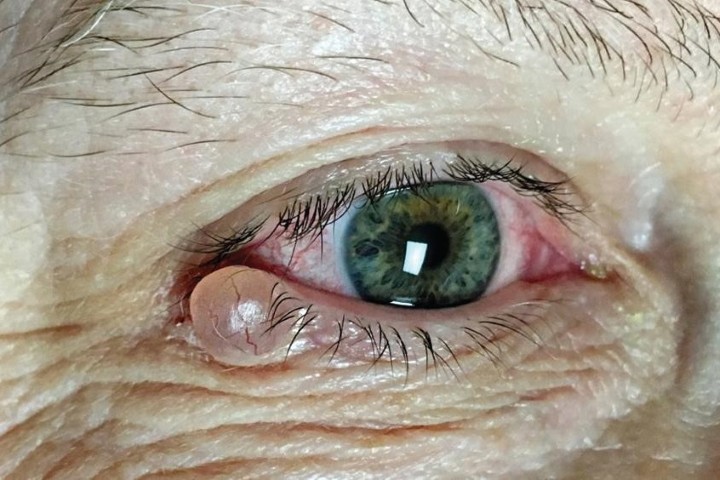
Conjunctival melanoma
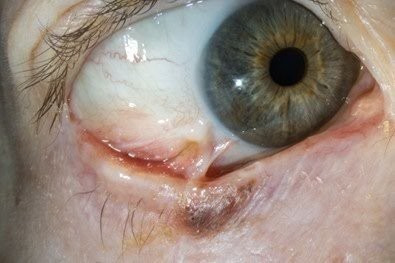
Conjunctival melanoma is uncommon and most pigmented conjunctival lesions encountered in practice are benign. Conjunctival naevi are the most common and typically present in the first two decades of life within the interpalpebral fissure. They often contain cysts, may enlarge slowly and generally do not require treatment. Racial melanosis presents as diffuse pigmentation in individuals with darker skin types and carries low malignant potential.
New or changing conjunctival pigmentation, particularly if nodular, irregular, vascularised or located in high-risk areas such as the plica, caruncle or fornices, should raise suspicion for conjunctival melanoma and prompt referral2.
Management relies heavily on appropriate initial treatment. Standard care involves wide local excision using a no-touch technique with adjunctive cryotherapy, often combined with mapping biopsies to determine disease extent. In practice, many patients are referred to ocular oncologists following incomplete excision elsewhere, frequently with positive margins. Given the importance of accurate staging and the impact of the first surgery on outcomes, early referral of suspicious lesions is strongly recommended.
Recurrence remains relatively common and adjuvant treatments such as topical mitomycin C or radiotherapy are often required. Given the molecular similarities between conjunctival and cutaneous melanoma, targeted systemic therapies, including BRAF and KIT inhibitors, may be considered in selected cases and can play an important role in disease control, sometimes allowing avoidance of more disfiguring procedures such as exenteration3.
Iris melanoma
Iris melanocytic lesions, most commonly naevi, are frequently encountered and typically present as well-defined pigmented lesions. Importantly, naevi and melanomas can appear very similar clinically, making differentiation challenging. While melanomas may present as discrete lesions, they can also be more subtle, with diffuse involvement associated with heterochromia, secondary glaucoma, recurrent uveitis, or spontaneous hyphaema.
Despite this, the prognosis is generally favourable, with a low metastatic risk and an iris melanoma–related mortality of approximately 5% at five years following diagnosis4. Careful longitudinal assessment is therefore essential. Serial anterior segment photography allows detection of change over time and any lesion demonstrating growth, elevated intraocular pressure or other concerning features should prompt referral.
A key consideration is distinguishing iris from ciliary body melanoma, since the latter carries a significantly higher risk of metastasis. Imaging plays an important role: anterior segment OCT can assist in measuring tumour thickness, but ultrasound biomicroscopy is critical for evaluating posterior involvement.
2024
Many iris melanomas can be safely observed. When treatment is required, options include surgical excision or radiotherapy. In many centres, plaque brachytherapy or proton beam therapy is used as primary treatment, particularly for lesions with ciliary body involvement, offering effective tumour control with low recurrence rates.
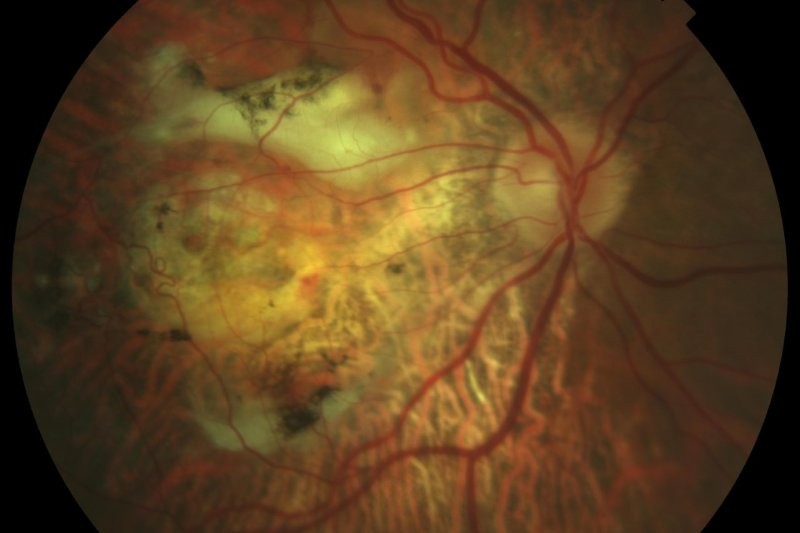
Choroidal melanoma
Choroidal melanoma is the most common primary intraocular malignancy and carries a significant risk of metastatic disease. Lesions at the posterior pole are more likely to present earlier due to visual symptoms, whereas peripheral tumours often remain asymptomatic. Up to 40% of patients are asymptomatic at diagnosis, often presenting with larger, more advanced tumours1.
This highlights the critical role of both optometrists and ophthalmologists. However, routine dilation is not consistently performed in clinical practice, influenced by patient factors, time pressures and underappreciation of its importance. Even within ophthalmology, not all consultations routinely include fundus examination. There are numerous examples of delayed diagnosis in patients followed long-term without posterior segment assessment, reinforcing the need for greater consistency in examination practices.
Distinguishing a benign choroidal naevus from melanoma is a common challenge. The MOLES scoring system (see Tables 1 and 2) provides a practical framework for risk stratification, guiding referral without the need for ultrasound5. Low-risk lesions can be monitored, while higher-risk lesions should be referred promptly.
Management continues to be guided by the Collaborative Ocular Melanoma Study (COMS), which demonstrated no survival advantage of enucleation over plaque brachytherapy6. As a result, eye-conserving treatments are now standard. In New Zealand, plaque brachytherapy remains the most commonly utilised modality, while stereotactic radiotherapy offers a non-surgical alternative with higher rates of visual complications.
Emerging therapies are expanding treatment options. Belzupacap sarotalocan (Bel-sar, previously known as AU-011), currently in phase 3 trials, represents a targeted therapy inducing tumour necrosis following light activation. Neoadjuvant agents such as darovasertib are also under investigation to preserve vision and reduce enucleation rates.
Despite advances in local tumour control, survival outcomes have not significantly improved, largely due to early micrometastasis7. Prognostication has therefore become increasingly sophisticated. In addition to the American Joint Committee on Cancer’s guidance on tumour staging, molecular profiling, including chromosomal alterations and gene expression profiling, plays a key role, with PRAME (preferentially expressed antigen in melanoma) expression further refining metastatic risk.
There is also increasing interest in less invasive methods of detecting systemic disease. Circulating tumour cells show promise for earlier detection of metastasis, although these technologies are not yet commercially available.
Systemic treatment of metastatic uveal melanoma remains challenging. Unlike cutaneous melanoma, immune checkpoint inhibitors have shown limited efficacy. However, newer therapies are beginning to improve outcomes. Tebentafusp has demonstrated a survival benefit in HLA-A*02:01-positive patients, while combination targeted therapies such as darovasertib with crizotinib have shown encouraging results in early clinical trials8,9.
Conclusion
While a growing number of novel therapies are emerging, these advances remain, in many ways, an ambulance at the bottom of the cliff. The most meaningful impact on outcomes still comes from early detection.
For optometrists and ophthalmologists, this means maintaining a high index of suspicion, performing thorough posterior segment examination, including routine dilation, and referring suspicious lesions early to subspecialist centres. Ultimately, earlier detection is what most improves both visual and life outcomes in ocular melanoma.
References
Dr Riyaz Bhikoo is a New Zealand-trained ophthalmologist with subspecialty expertise in vitreoretinal surgery, medical retina, ocular oncology, and cataract surgery. He practises at Auckland Eye and Greenlane Clinical Centre. He completed advanced fellowships in Australia, Canada, and the US and now leads the plaque brachytherapy service for ocular melanoma in New Zealand, while remaining actively involved in clinical research and collaborative care.